The etiology of this disease is still unclear (Moller 2017). It is hypothesized that one
or more unidentified antigens induce an immune response mediated by alveolar
macrophages and lymphocytes. Due to similarities with granulomas formed
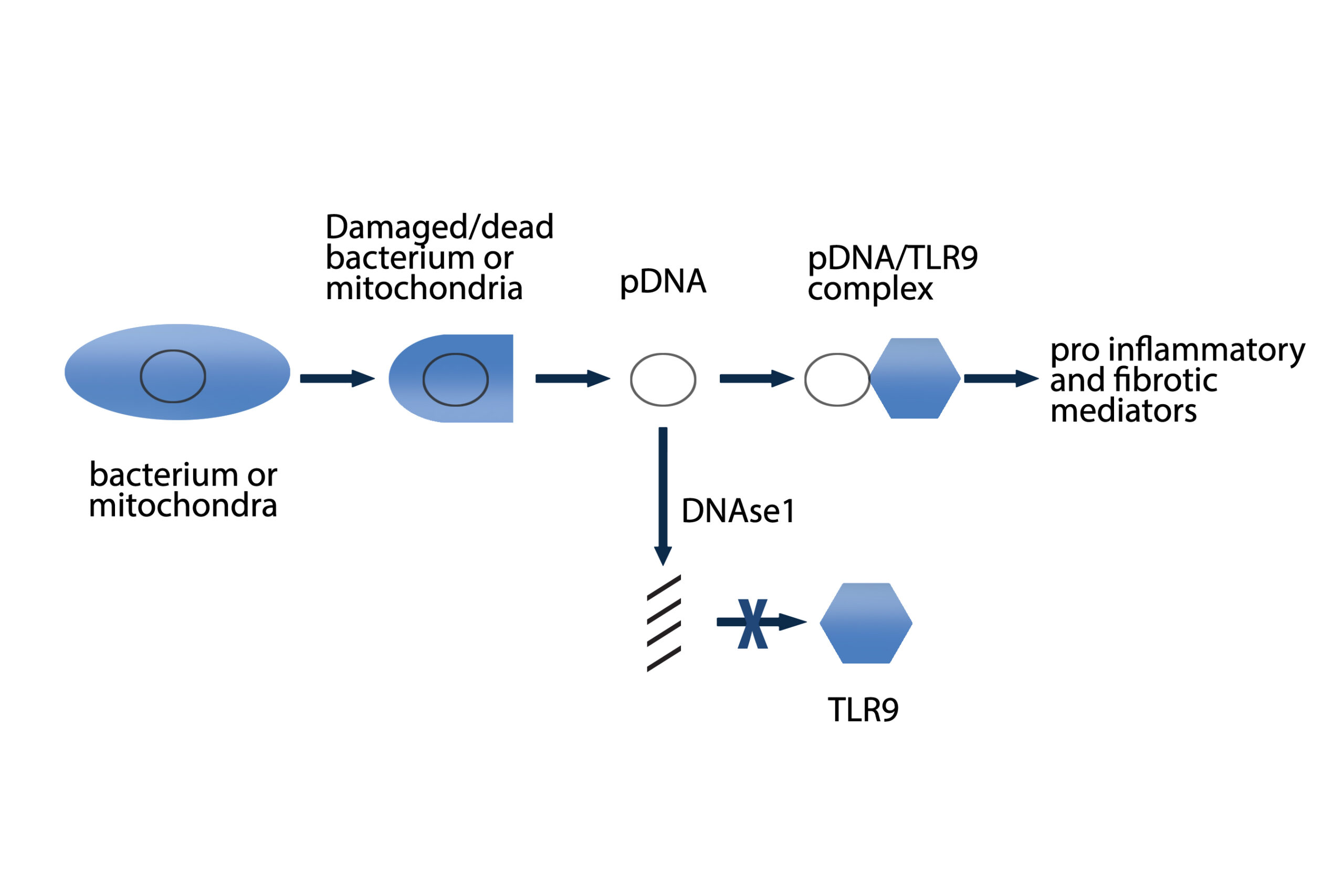
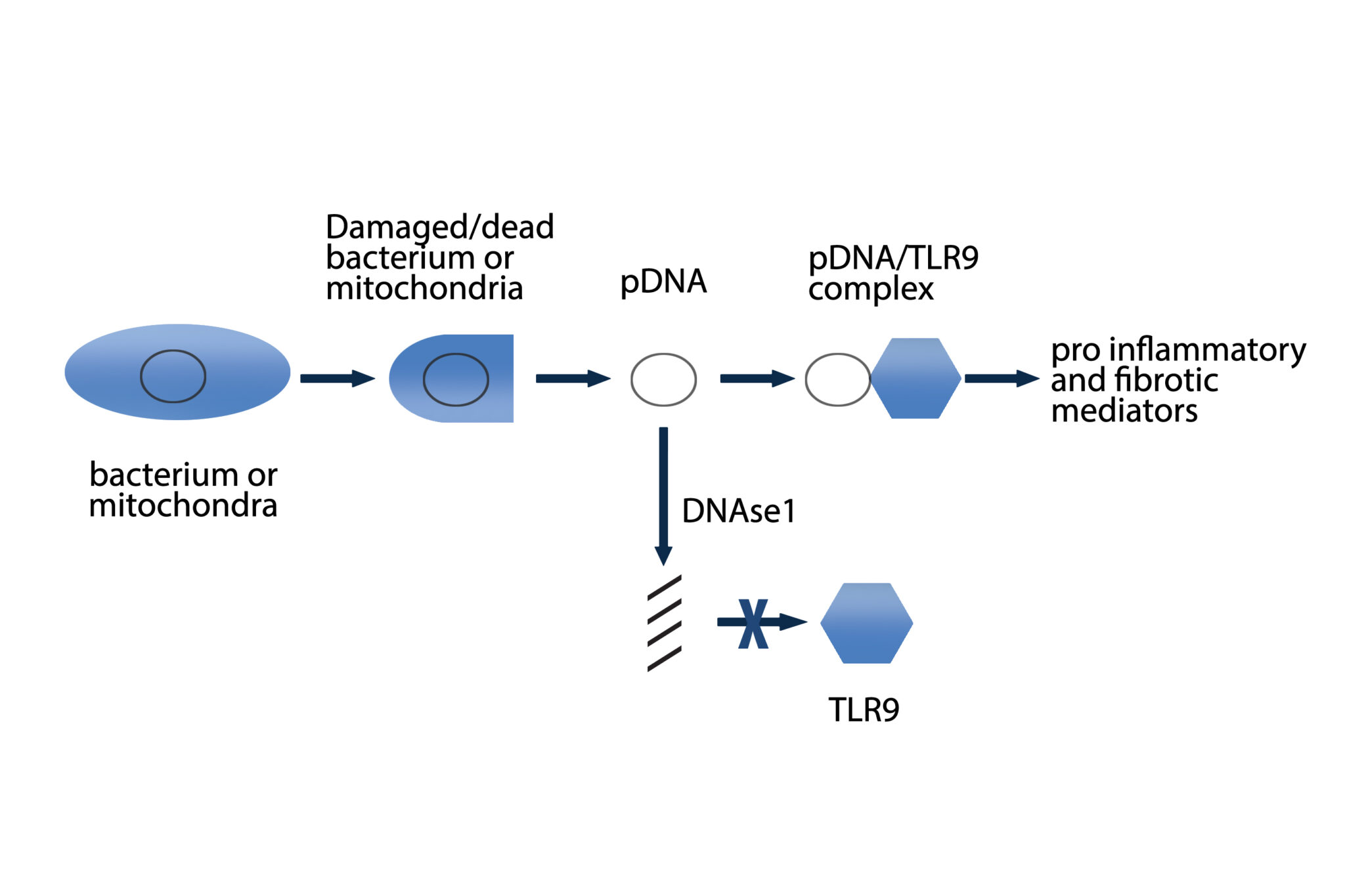
during mycobacterial infections, it is hypothesized that DNA containing complexes originating from a variety of bacteria may be one of these potential antigens
(Eishi 2002; Gupta 2007; Drake 2013). There is evidence of the presence of microbial DNA from mycobacteria, propionibacteria and borrelia in sarcoidosis tissues
suggesting they may participate in sarcoidosis granuloma formation (Chen and
Moller 2014). Bacterial amyloid curli may act as a carrier for such DNA to elicit an
autoimmune response (Tursi 2017; Nicastro and Tukel 2019). Serum amyloid A
derived from the host may participate in the granuloma formation by regulating
inflammation through Toll-like receptor-2 (TLR2) (Chen 2010). Serum amyloid A
localized to macrophages and giant cells within sarcoidosis granulomas but
correlated with CD3(+) lymphocytes, linking expression to local Th1 responses
(Chen 2010). Serum amyloid A activated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappaβ) in TLR2-expressing human cell lines, regulated experimental Th1-mediated granulomatous inflammation through IFN-γ, TNF, IL-10, and TLR-2; and stimulated production of tumor necrosis factor, IL-10, and IL-18 in lung cells from patients with sarcoidosis, effects inhibited by blocking TLR-2 (Chen 2010). A significant association was found between TLR9 expression and CD4+ lymphocytes in bronchoalveolar lavage (BAL) of patients with sarcoidosis. Increased TLR9 expression in alveolar macrophages derived from patients with sarcoidosis was also reported and these cells secreted higher levels of cytokines in response to in vitro stimulation (Inaoka 2019). Additional antigens implicated in initiating the inflammatory cascade leading to granuloma formation include beta-actin, intermediate filament vimentin and pathogen-associated molecular patterns (PAMPs) (Cooke 2013; Mortaz 2017).
The granuloma in sarcoidosis is characterized by a core of monocyte-derived epithelioid histiocytes and multinucleate giant cells with interspersed CD4+ T lymphocytes (Iannuzzi 2007). A minority of cells in or near the granuloma are CD8+ T lymphocytes, fibroblasts, regulatory T cells, and B lymphocytes. The T-cell response is biased toward a Th1 phenotype, with important roles for IFN-γ and IL-12 (Moller 1996). Increased expression of key inflammatory mediators, TNF, IFN-γ, IL-2, IL-10, IL-12, IL-18 and transcription factor STAT-1, is indicative of a Th1 mediated immune response (Baughman 2011). More recently, studies have found upregulation of Th17 immune responses producing IL17 and Th17.1 responses expressing INFg in sarcoidosis (Facco 2011; Ramstein 2016). Several studies indicate an expansion of regulatory T cells but with reduced functional capabilities that may be ineffective in suppressing the local exaggerated T cell mediated inflammatory tissue responses (Miyara 2006; Taflin 2009). Sarcoidosis has paradoxical effects on inflammatory processes, characterized by increased macrophage and CD4 helper T cell activation, resulting in accelerated inflammation, but immune response to antigen challenges such as tuberculin is suppressed in the peripheral blood and skin. This response may be mediated by upregulation of the immune checkpoint inhibitor Programmed Death-1 (PD-1) on peripheral blood CD4 T cells in sarcoidosis patients (Celada 2017).
{kind=link}